

Product Verification (By way of verification)
Product Verification:The Product Verification (By way of Verification) process, accordance to Circular Letter of the Medical Device Authority No. 2 Year 2014 (Ref.: (5).dlm.MDA.100-1/8/5) - dated 22 May 2014, allows for expedited registration of medical devices that have already been approved in other jurisdictions with recognized regulatory systems, as of date, those approval recognised by MDA.
New registration as set out in HOW TO APPLY FOR MEDICAL DEVICE REGISTRATION UNDER MEDICAL DEVICE ACT 2012 (ACT 737), MDA/GL/MD-01, June 2014, Second Edition
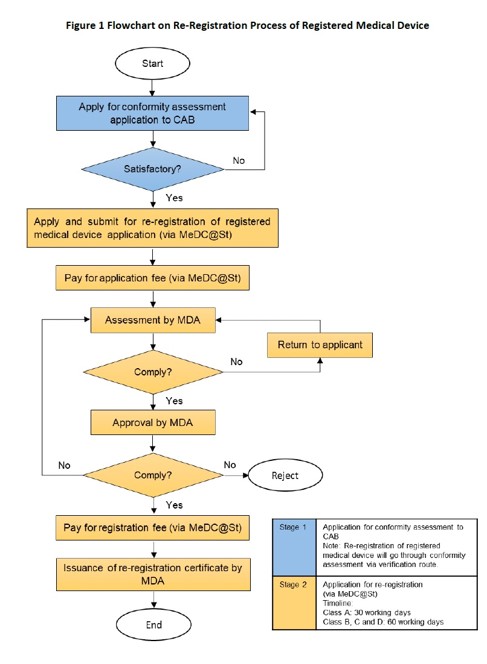
Re-registration process as set out in GUIDELINE FOR RE-REGISTRATION OF REGISTERED MEDICAL DEVICE, MDA/GL/08, September 2022, Second Edition
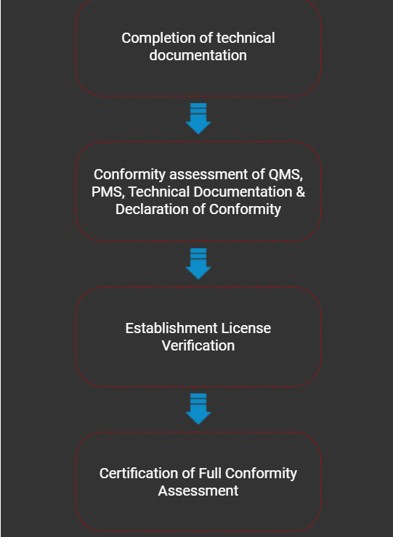
Medical devices which have not obtained any approval by regulatory authorities or notified bodies listed in Table 1 is required to undergo full conformity assessment by any registered CAB in accordance with the requirements stipulated in Section 7(1)(a) of Act 737.
Full Conformity Assessment
The Full Conformity Assessment (FCA) by the Medical Device Authority (MDA) in Malaysia is a comprehensive evaluation process for medical devices that ensures they meet regulatory requirements for safety, quality, and effectiveness before being registered and marketed. It is applicable to all medical devices that have been subjected to conformity assessment and approved by any of the recognised foreign regulatory authorities or notified bodies with respective approval type as shown in Table 1 accordance to Circular Letter of the Medical Device Authority No. 2 Year 2014 (Ref.: (5).dlm.MDA.100-1/8/5) - dated 22 May 2014, except for those which are exempted from registration in Malaysia.
What is Conformity Assessment?
Conformity assessment is a systematic and ongoing examination of evidence and procedures to ensure the safety, performance, benefit and risk of medical devices. It is also to ensure manufacturing compliance to essential principles of safety and performance (EPSP) and requirements of the Medical Device Act 2012 (Act 737). The classification of a medical device determines the conformity assessment procedures to be undertaken. Conformity assessment becomes more stringent as the risk of medical device increases Conformity Assessment (Product Verification) and Full conformity Assessment.
Elements of conformity assessment
For the purpose of medical device registration, the following conformity assessment elements shall be assessed by the CAB:
a) Quality management system (QMS) - Manufacturers shall be certified with ISO 13485.
Extend of QMS certification based on risk class
- Risk Class A and B.
the quality management system for a Class A or Class B device shall be either a full quality management system or one without design and development control, the manufacturer shall choose the one that it believes to be most suitable.
- Risk Class C and D.
Establish and maintain a full QMS
b) A system for post-market surveillance;
1) For Class B, C and D medical device, CAB shall ensure PMS is established, maintained and implemented by the establishment.
2) The following processes shall be documented, maintained and implemented by the establishment:
a) complaint handling;
b) distribution records;
c) mandatory problem/adverse event reporting;
d) field corrective action; and
e) recall.
f) Technical documentation; and
- i) The technical documentation provides the evidence that the medical device meets the essential principles for safety and performance (EPSP). The manufacturer shall collect and examine evidences of conformity and compile these evidences in a technical documentation.
- ii) The manufacturer shall also establish a summary of the technical documentation in the form of a common submission dossier template (CSDT), which is to be held or submitted, as required depending on the class of the medical device.
- iii) CAB determines the adequacy of the documented evidence in support of the manufacturer’s declaration of conformity to the EPSP through a review of the CSDT and technical documentation.
- i. CAB shall refer to ISO 16142-1 and ISO 16142-22 for selection of standards in support of EPSP. CAB shall also identify relevant established standards applicable for the medical devices within the technical area(s) they are registered for. Use of established standards to demonstrate the compliance with EPSP shall be according to the following priority:
a) Malaysian Standards (MS) and/ or International Standards (i.e. ISO/IEC/ITU);
b) Recognised Countries Standards, i.e. US, EU, Canada, Australia and Japan; and
c) Industry Standards/ Company standards.
In the case the manufacturer opts to diverse from the above hierarchy, the manufacturer shall provide valid justification for its deviation.
d) A declaration of conformity.
- i) Medical Devices manufacturers shall attest that its medical device complies fully with all applicable Essential Principles for Safety and Performance and other requirements of Act 737 and the subsidiary regulations under it, documented in a written 'Declaration of Conformity' (DOC).
- ii) Guidance Document MDA/GD/0025 specifies requirements on Declaration of Conformity and shall be used for this purpose.
- iii) The CAB shall ensure that all elements are clearly stated and/or listed and the DoC has been signed by a person from the top management of the manufacturer or a person authorised to sign on his/her behalf.
a) Extend of QMS certification based on risk class
- 3) Risk Class A and B
the quality management system for a Class A or Class B device shall be either a full quality management system or one without design and development control, the manufacturer shall choose the one that it believes to be most suitable.
- 4) Risk Class C and D
Establish and maintain a full QMS
Section 5 (1) of Medical Device Act 2012 (Act 737) requires a medical device to be registered under the Act before it can be imported, exported or placed in the market. For that purpose, an application for the registration of a medical device must be made according to the requirement under Act 737 and in the manner determined by the Authority in Medical Device Regulation 2012.
Starting from 1 July 2013 when Act 737 comes into effect, all medical devices to be placed market are required to be registered under the Act. The application for medical device registration shall be made to the Authority through an online, web-based system called “Medical Device Centralized Online Application System (MeDC@St)".
The term “medical device” covers any product used in healthcare for the diagnosis, prevention, monitoring or treatment of illness or handicap but excludes drugs.
The persons responsible for registering a medical device under Act 737 are—
(i) the manufacturer of medical device as defined in Section 2 of Act 737; and
(ii) in the case of a medical device manufactured in foreign country, the authorized representative of the foreign manufacturer, as defined in Section 2 of Act 737
- (1) It is applicable to all medical devices that have been subjected to conformity assessment and approved by any of the recognised foreign regulatory authorities or notified bodies with respective approval type as shown in Table 1, except for those which are exempted from registration in Malaysia.
Table 1: Recognised foreign regulatory authorities and notified bodies and the respective approval types eligible for conformity assessment by way of verification process
|
Recognised foreign regulatory authority or notified body |
Approval Type |
|
(i) Therapeutic Goods Administration (TGA) Australia |
TGA licence |
|
(ii) Health Canada, Canada |
Health Canada medical device licence |
|
(iii) Notified bodies listed in New Approach Notified and Designated Organisations (NANDO) database of European Union (EU) |
For general medical device: • Annex 11 Section 3 or Annex V of MOD (for Class IIA) • Annex II Section 3 or Annex Ill coupled with Annex V of MDD (for Class 118) • Annex II Section 3 and 4 of MDD (for Class 111) • Annex II Section 3 and 4 of AIMDD (for active implantable medical device) For IVD medical device: • Annex IV (Including Section 4 and 6) of IVDD (for List AIVD) |
|
Recognised foreign regulatory authority or notified body |
Approval Type |
|
• Annex IV (excluding Section 4 and 6) or Annex V coupled with Annex VII of IVDD (for List B and self- testing IVD) • Annex Ill, EC declaration of conformity (Section 1 to 5 of Annex Ill). Applicable for only Class B IVD medical device in accordance with Medical Device Reoulation 2012. |
|
|
(iv) Ministry of Health, Labour and Welfare (MHLW) Japan |
• Pre Market Certification from a Japanese Registered Certification Body (RCB and PMDA). • Pre Market Aooroval from MHLW. |
|
(v) Food and Drug Administration (FDA), United States of America (USA) |
• US FDA 510(k) clearance letter • US FDA pre-market approval (PMA) letter |
|
(vi) Other foreign regulatory authorities or notified bodies |
To be determined by MDA from time to time |
- (2) Medical devices which have not obtained any approval by regulatory authorities or notified bodies listed in Table 1 is required to undergo full conformity assessment by any registered CAB in accordance with the requirements stipulated in Section 7(1)(a) of Act 737.
B. Legal Basis
- (3) Section 7(1)(a) of Act 737 prescribes that upon receipt of an application made under Section 6 of the Act, MDA being satisfied that the medical device has been subjected to the conformity assessment procedures to be carried out by the CAB, MDA may register the medical device for a prescribed period.
- (4) Section 10(1) of Act 737 prescribes that a CAB shall be a body registered under this Act to carry out conformity assessment of a medical device to be registered.
- (5) For the purpose of product registration, it is further required that all medical devices shall be subjected to conformity assessment to demonstrate its conformity to the requirements as specified in Third Schedule of Medical Device Regulation (MOR) 2012.
C. Eligibility of CABs to conduct verification process
- (6) Section 10 of Act 737 prescribes that conformity assessment of medical device shall be carried out by CAB. Hence, it is pertinent that the CABs shall carry out the conformity assessment with due diligence and adhering to all the regulatory requirements as stipulated in Act 737 and its subsidiary legislations. The CABs shall be independent and impartial with regards to the performance of its conformity assessment duties as stipulated in Section 10(3)(a) of Act 737 and paragraphs 9(2) and 9(7) of Fourth Schedule of MOR 2012
- (7) The following criteria shall apply for the eligibility of CABs for performing conformity assessment of a medical device by way of verification process-
(i) For medical devices which have been subjected to conformity assessment by recognised foreign regulatory authorities in Table 1, the verification process of the said medical device may be carried out by any CABs registered under Act 737;
(ii) Where a medical device has its conformity assessment already carried out by a recognised foreign notified body in Table 1, then the subsidiary of the foreign notified body registered as CAB under Act 737 shall not be allowed to do verification on the same medical device. However, it may be carried out by any other CABs registered under Act 737;
(iii) Where a medical device has its conformity assessment already carried out by any other regulatory authorities recognised by MDA from time to time, then the criteria in (ii) shall apply; and
(iv) The CABs which are eligible to conduct verification process shall have been registered with at least one scope under Medical Device Technical Area (Appendix 1 of Fourth Schedule MOR 2012). Example: A CAB with registered scope of MD 0203 may conduct verification process on a medical device under any Medical Device Technical Areas.
D. Conformity assessment elements and parameters to be verified by CAB
- (8) The parameters to be verified by CAB in its verification process shall comprise of the conformity assessment elements as stipulated in Third Schedule of MOR 2012 as follows-
- (i) Conformity assessment of quality management system;
- (ii) Conformity assessment of post market surveillance system;
- (iii) Conformity assessment of technical documentation; and
- (iv) Conformity assessment of declaration of conformity.
- (9) The extent of verification activities will depend on the class of the medical device.
E. Verification steps for Class A (active, sterile or with measuring function) medical device
- (10) Verification steps and parameters to be verified by CAB for Class A (active, sterile or with measuring function) medical device are described in Figure A and Table 2 respectively.
Figure A: Verification steps for Class A (active, sterile or measuring function) medical
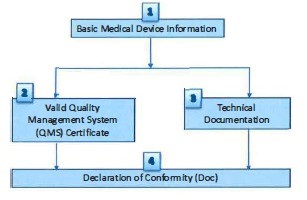
Table 2: Verification steps and parameters to be verified for Class A (active, sterile or measuring function) medical device
|
Step |
Parameters to be verified |
|
1) Basic Medical Device information |
(i) It shall be a medical device, based on intended use as described in technical documentation; (ii) The intended use/indication for use shall be the same as what has been approved by the recognised country; (iii) Classification and application of risk classification rule shall be in accordance with Appendix 1of First Schedule MOR 2012; and (iv) Grouping of medical device shall be in accordance with Rule of Grouping in Second Schedule MOR 2012. |
|
2) Valid Quality Management System (QMS) Certificate |
(i) Authenticity of the manufacturer's OMS certificate, eg ISO 13485 or other equivalent QMS certificate, issued by foreign recognised notified body or regulatory authority granting the certificate; (ii) Scope of QMS of the manufacturer of medical device as required by Third Schedule of MOR 2012; and (iii) All certificates submitted shall be within validity period. |
|
3) Technical documentation |
(i) Any claims of intended use on labels and labelling shall be consistent with information on intended use provided in step 1. ii) Labelling shall be in accordance with Sixth Schedule MOR 2012 iii) Aspects of manufacturing processes concerning- a. securing and maintaining sterile conditions, eg sterilization report stating that packaging material and expiry date achievable with the method of sterilization employed; b. conformity with metrological requirements, eg calibration certificate; c. conformity with active medical device requirements, eg IEC 60601 test certificate (test report shall be made available upon |
|
Step |
Parameters to be verified |
|
request by CAB and MDA). |
|
|
4) Declaration of conformity (DoC) |
DoC including supporting documents prepared according to Appendix 3 Third Schedule MOR 2012. |
Table 2. Conformity assessment on Class A medical devices
|
Conformity assessment element |
Manufacturer/AR responsibility |
Conformity assessment requirement |
Clause |
|
Quality Management System |
a) Establish and maintain a full QMS or may exclude design and development |
Premarket and regulatory audit not required. |
5.1. |
|
(QMS) |
controls, process control and inspection and testing; and /or b) Keeping evidence on the aspect of manufacture concerned with: i. Securing and maintaining sterile condition if the medical device is to be supplied sterile: Validation report and/ or; ii. Conformity of the medical device with the metrological requirements if the medical device has a measuring function: Calibration report/certificate. |
||
|
Post-market surveillance system |
a) Establish and maintain PMS system b) Record and evaluate reports of adverse events. c) document, maintain and implement: i. complaint handling; ii. distribution records; iii. mandatory problem/adverse event reporting; iv. field corrective action; and v. recall. |
Premarket and regulatory audit not required. Regulatory audits may be conducted on establishment as deemed necessary by the Authority. |
5.2. |
|
Technical Documentation |
Prepare summary of technical documentations in the format of CSDT (refer to MDA/GD/0008: CSDT) and have available for review upon request. |
Premarket submission of CSDT not required. May be requested by the Authority for the purpose of investigating specific safety or regulatory concerns. |
5.3. |
|
Declaration of Conformity (DoC) |
Prepare DoC as per specified in MDA/GD/0025. |
Manufacturer/ AR to submit to Authority during registration process and to keep in file and present upon request by the Authority. |
5.4. |
F. Verification steps for a Class B, C or D medical device
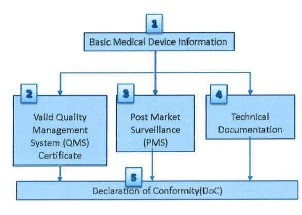
- (11) Verification steps and parameters to be verified by CAB for a Class B, C or D medical device are described in Figure Band Table 3 respectively.
Figure 8: Verification steps for a Class B, C or D medical device
|
Step |
Parameters to be verified |
|
1) Basic Medical Device Information |
(i) It shall be a medical device, based on intended use as described in technical documentation; (ii) The intended use/indication for use shall be the same as what has been approved by the recognised country; (iii) Classification and application of risk classification rule shall be in accordance with Appendix 1of First Schedule MOR 2012; and (iv) Grouping of medical device shall be in accordance with Rule of Grouping in Second Schedule MOR 2012. |
|
Step |
Parameters to be verified |
|
2) Valid QMS Certificate |
(i) Authenticity of the manufacturer's QMS certificate, eg ISO 13485 or other equivalent QMS certificate, issued by foreign recognised notified body or regulatory authority granting the certificate; (ii) Scope of QMS of the manufacturer of medical device as required by Third Schedule of MOR 2012; and (iii) All certificates submitted shall be within validity period |
|
2) Post-market surveillance (PMS) |
(i) List of reported ongoing incident globally (if applicable); (ii) List of incidents that have been resolved for the past 3 years (if applicable); and (iii) Date of last audit. |
|
3) Technical documentation |
(i) Authenticity and validity of CE mark certificate and/or evidence of approval by recognised foreign regulatory authority; (ii) Common Submission Dossier Template (CSDT); (iii) Any claims of intended use on labels and labelling shall be consistent with information on intended use provided in step 1; (iv) Labellinq shall be in accordance with Sixth Schedule MOR 2012, |
|
4) Declaration of Conformity (DoC) |
DoC including supporting documents prepared according to Appendix 3 Third Schedule MOR 2012. |
Table 3. Conformity assessment on Class B medical devices
|
Conformity assessment element |
Manufacturer/AR responsibility |
Conformity assessment requirement |
Clause |
|
Quality Management System (QMS) |
a) Establish and maintain a full QMS or may exclude design and development controls, process control and inspection and testing; and b) Appoint CAB to review and conduct on- site audit if necessary, to verify evidence of conformity to QMS requirements. |
CAB shall be satisfied that a current and appropriate QMS is in place or otherwise conduct a QMS audit on manufacturer prior to certification. |
5.1. |
|
Post Market Surveillance |
a) Establish and maintain PMS system. |
CAB shall ensure an appropriate post-market |
5.2 |
|
b) Record and evaluate reports of adverse events. c) document, maintain and implement: i. complaint handling; ii. distribution records; iii. mandatory problem/adverse event reporting; iv. field corrective action; and v. recall. |
surveillance system is established, maintained and implemented by manufacturer/ AR. |
||
|
Technical Documentation |
a) Collect and examine evidence and undertake procedures to determine conformity of medical device to EPSP; b) Prepare summary of technical documentations in the format of CSDT (refer to MDA/GD/0008: CSDT). |
CAB to conduct a review of the CSDT to verify evidence of compliance with EPSP requirements. |
5.3. |
|
Declaration of Conformity (DoC) |
Prepare DoC as per specified in MDA/GD/0025. |
CAB to review and verify adequacy of the DoC. |
5.4. |
Table 4. Conformity assessment on Class C and D medical devices
|
Conformity assessment element |
Manufacturer/AR responsibility |
Conformity assessment requirement |
Clause |
|
Quality Management System (QMS) |
Establish and maintain a full QMS and appoint CAB to review and assess QMS compliance. |
Be satisfied that a current and appropriate QMS is in place or otherwise conduct a QMS audit on manufacturer prior to certification. |
5.1. |
|
Post Market Surveillance |
a) Establish and maintain PMS system. b) Record and evaluate reports of adverse events. c) document, maintain and implement: i. complaint handling; ii. distribution records; iii. mandatory problem/adverse event reporting; iv. field corrective action; and v. recall. |
CAB shall ensure an appropriate post-market surveillance system is established, maintained and implemented by manufacturer/AR |
5.2. |
|
Technical Documentation |
a) Collect and examine evidence and undertake procedures to determine conformity of medical device to EPSP; b) Prepare summary of technical documentations in the format of CSDT (refer to MDA/GD/0008: CSDT). |
CAB to conduct a review of the CSDT to verify evidence of compliance with EPSP requirements |
5.3. |
|
Declaration of Conformity (DoC) |
Prepare DoC as per specified in MDA/GD/0025. |
CAB to review and verify adequacy of the DoC. |
5.4. |
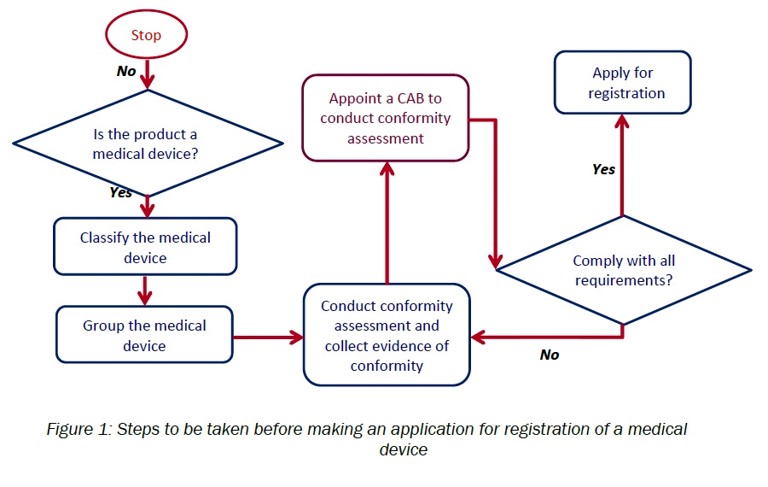
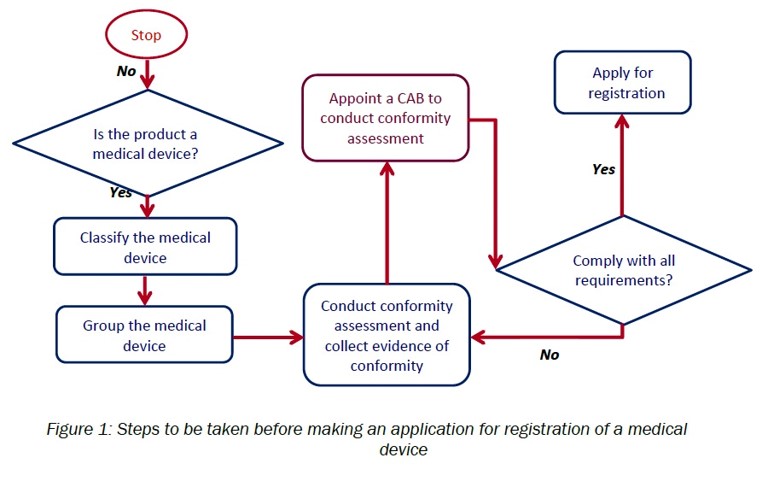
G. Medical device registration:
What are the steps and criteria for medical device registration?
- Determine whether the product is a medical device
The determination of the product will be based on the definition of “medical device” in as specified in Section 2 of Act 737 and further elaborated in the Guidance Document on Definition of Medical Device (MDA/GD-01).
- Appropriately classify the medical device
The classification of medical device should be done according to the rules of medical device classification as specified in First Schedule of Medical Device Regulation 2012 and further elaborated in the Guidance Document on The rules of Classification for general medical devices (MDA/GD-04).
- Appropriately group the medical device
The grouping of medical device should be done according to the rules of medical device grouping as specified in Second ScheduleofMedical Device Regulation 2012 and further elaborated in the Guidance Document on Grouping of Medical Device.
- Conduct conformity assessment and collect evidence of conformity
(i) the evidence of conformity has to be collected to demonstrate compliance to applicable Essential Principles of Safety and Performance of Medical Device as specified in Appendix 1 of Third Schedule of Medical Device Regulation 2012 and further elaborated in the Guidance
Document on Essential Principles of Safety and Performance of Medical Device (MDA/GD-02);
(ii) the evidence of conformity has to be compiled according to the Common Submission Dossier Template (CSDT) as specified in Appendix 2 of Third Schedule of Medical Device Regulation 2012 and further elaborated in the Guidance Document on Common Submission Dossier Template (MDA/GD- 03);
(iii) the Declaration of Conformity according to the template in Appendix 1A of Third Schedule of Medical Device Regulation 2012 has to be duly prepared, signed and
stamped.
- Appoint CAB to conduct conformity assessment
According to 3rd Schedule of Medical Device Regulation 2012
(i) the evidence of conformity has to be verified or validated by the registered CAB;
(ii) the CAB has to issue certificate of conformity and the report upon completion of the conformity assessment.
- Apply to register medical device using MeDC@St
(i) Application for registration of medical device may be made after the criteria are met and the information and supporting documents to support the criteria are available;
(ii) Application for medical device registration shall be made via MeDC@St;
(iii) Applicant must create an account before making application via MeDC@St.
After the account have been created in the MedC@st system, the form need to be filled in and it consist of 8 parts as stated below:
(i) General information;
(ii) Information of manufacturer;
(iii) Grouping of medicaldevice;
(iv) Common Submission Dossier Template (CSDT);
(v) Supporting documents forCSDT;
(vi) Post-market vigilance history;
(vii) Declaration of Conformity
(viii) Attestation for medical device registration application
How to complete the form:
- 1. Is the medical device for export only
- 2. Is the medical device contains any active ingredient, poison or drug?
- 3. Type of medical device
- 4. Class of medical device
- 5. Classification rules
- 6. Medical device category
- 7. Medical device name
- 8. Description of medical device
- 9. Information on the product formulation (applicable for medical device contains the active ingredient/poison/drug)
- 10. Intended use of the medical device
- 11. HS code
Please provide the HS Code for the medical device, if applicable. HS Code is Harmonized Tariff Nomenclature & Coding System which was created for international use by
the Custom Department to classify commodities when they are being declared at the custom frontiers by exporters and importers. For reference of HS Codes, you may search from
Search Tariff function at JKDM HS
– Explorer Website at http://tariff.customs.gov.my.
– Download Checklist for medical device conformity assessment by CAB
For more info, please visit http://www.customs.gov.my.
- 12. GMDN code
- 13. Premarket clearance
- 14. Conformity assessment done by CAB
- 15. Information of Manufacturer
- 16. Grouping of Medical Device
- 17. Medical device grouping
- a) Please select the grouping that is applicable to your device. The grouping should be done in accordance with the Rules of Grouping as specified in the Second Schedule of the Medical Device Regulation 2012 and further elaborated in the Guidance Document on Grouping of Medical Device.
- b) Same manufacturer. Please specify whether or not constituent-components or medical devices that are grouped together are manufactured by the same manufacturer.
- c) List of Constituent-components medical devices. Please list the constituent-components or medical devices that are grouped together in fields provided. You may list all in a template provided in the link for batch uploading.
- d) Common Submission Dossier Template (CSDT)
- e) Post-Market vigilance history
- f) Declaration of Conformity
H.For EC Certificate MDA APPROACH FOR NEW AND RE-REGISTRATION OF MEDICAL DEVICES WITH EXPIRED EC CERTIFICATES AND SELF-DECLARED CLASS B IVD:
- Contact for more details:
Email: fleming@cciglobe.com, reecheeteo@cciglobe.com
Tel:+(603) 5131 6160